PELE Molecular Parameters¶
The parameters for the force fields used in PELE, the rotamer libraries used during side chain prediction (and the side chain exploration done when needed during the perturbation), and conformation libraries used during conformation perturbation (when applicable) are located in the Data/ directory or, for non-standard molecules and residues, in your DataLocal/ directory. In particular:
<DataDir>/Templates/<forcefield>/HeteroAtoms/contains the force field parameters for non-protein and non-nucleic acid residues (ligands, water molecules, etc.).<DataDir>/Templates/<forcefield>/DNA/contains the force field parameters for DNA residues.<DataDir>/Templates/<forcefield>/RNA/contains the force field parameters for RNA residues.<DataDir>/Templates/<forcefield>/Protein/contains the force field parameters for protein residues.<DataDir>/SGB/contains parameters for the solvent-related energy when using either VDGBNP or SGBNP (see solventType).<DataDir>/OBC/solventParamsHCTOBC.txtcontains parameters for the solvent-related energy when using OBC (see solventType).<DataDir>/LigandRotamerLibs/contains the rotamer libraries for non-protein and non-nucleic acid residues (usually ligands and cofactors).<DataDir>/RotamerLibs/contains the rotamer libraries for DNA, RNA and protein residues.<DataDir>/Conformations/contains the conformation libraries.
where <DataDir> refers to either Data/ or DataLocal/, and the <forcefield> directory is one of the currently supported ones (see ForceField).
If you use an hetero group not included in the data directories, then you should create and add both the template file with the force field parameters (under <DataDir>/Templates/<forcefield>/HeteroAtoms/), the OBC parameters for that group at the end of solventParamsHCTOBC.txt (only if you use the OBC solvent model), the rotamer library definition file (at <DataDir>/LigandRotamerLibs/) or the conformation library definition file (at <DataDir>/Conformations). The following sections explain how to generate the different data needed.
Solvent parameters¶
You only need to create solvent parameters if you are using OBC, because parameters for other solvent models are generic and already apply to any atom in your molecules (in SGB, the parameters are already included in the template file; check IMPACT template file format; also see Solvent parameters for SGB; for the penalty terms file format, check Solvent penalty terms).
OBC¶
To run a PELE simulation using the OBC implicit solvent model, you need the atomic parameters for all the atoms in your system. PELE already includes the parameters for the atoms of the residues appearing in a macromolecule (DNA or protein), and for some small molecules. If your molecule is not parameterized (PELE will complain with an error message), you can create the parameters in the following way:
$ cp /path/to/my/template mytemplate
$ python /path/to/PELE-1.5/scripts/solventOBCParamsGenerator.py mytemplate
You will then obtain a file named mytemplate_OBCParams.txt with a line per atom of your molecule, with the OBC parameters. Then, paste those lines at the end of PELE’s data file Data/OBC/solventParamsHCTOBC.txt or, even better, make a copy of the solventParamsHCTOBC.txt data file in your project’s directory, under ./DataLocal/OBC/solventParamsHCTOBC.txt, and paste the molecule parameters there, so that you don’t change your central data file, but instead use a local copy (PELE gives precedence to ./DataLocal/ files over those in ./Data/).
Notice that the parameters will be used whatever force field (OPLS2001, OPLS2005, etc.) you are using. The script uses atom types from mytemplate. As long as they are the same in all the force fields, you can use whichever you prefer. If types differ, but are not one of the recognized type names (see the script), then the parameters will be assigned depending on the atom name. Finally, if the atom name is not recognized either, then default parameters will be assigned. Even in the case of differing atom types depending on the force field, it may happen that the OBC parameters for the specific atom types be the same.
You will find further information in the documentation inside the PELE script scripts/solventOBCParamsGenerator.py.
Template parameters¶
You can find a description of the file formats used at:
IMPACT (standard template format): IMPACT template file format.
PELE Custom format (for inter-residue parameters): PELE custom template file format.
Non-ligand rotamer library: Non-ligand rotamer library file.
Ligand rotamer library: Ligand rotamer library file.
Conformation library: Conformation library file.
OPLS¶
You can create the template and rotamer library files from either a file in PDB format or in MAE format. In any case, it may be helpful to check with Maestro the topology and charges of your ligand.
In the following examples, we will assume Schrödinger software is installed under /opt/schrodinger/, and that PlopRotTemp.py is in /opt/pele/scripts/PlopRotTemp.py.
From a PDB file, run:
$ /opt/schrodinger/utilities/pdbconvert -ipdb myligand.pdb -omae myligand.mae
$ /opt/schrodinger/utilities/python /opt/pele/scripts/PlopRotTemp.py -OPLS=2005 myligand.mae
From a MAE file, first you have to make sure that your ligand has a PDB residue name assigned, and that all PDB atom names are unique. To do this:
Open you ligand in Maestro.
Select it.
Go to Build and under Other Edits select Change Other Properties.
Select property Residue / Chain Name.
Enter the 3-letter name you want for your residue (see Fig. 11).
Apply your changes.
Select property PDB Atom Name.
Mark option Set unique PDB atom names within residues (see Fig. 12).
Apply your changes and close the dialog.
Save your ligand in a new MAE file.
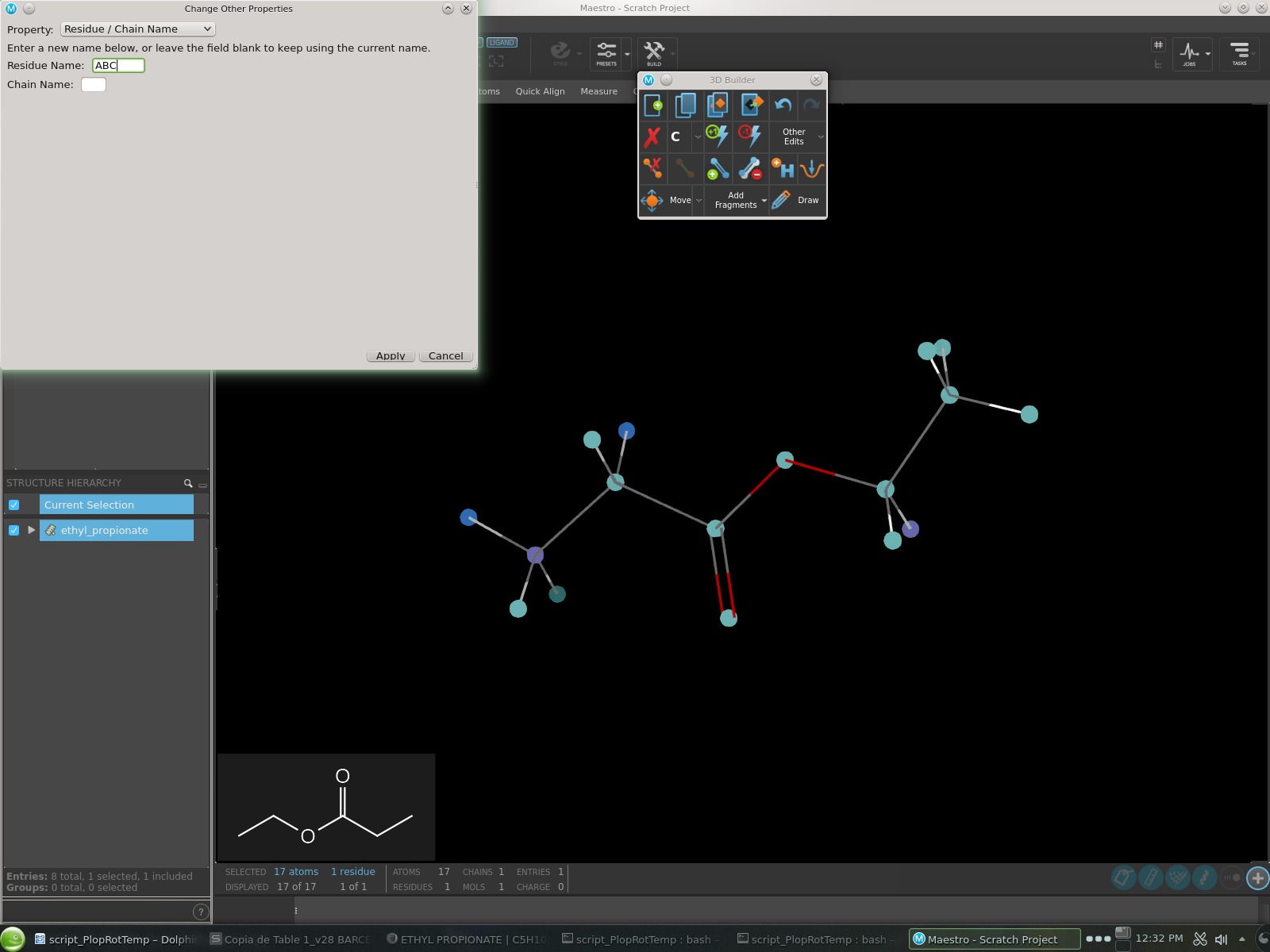
Fig. 11 Dialog to set the PDB residue name for your ligand in Maestro.¶
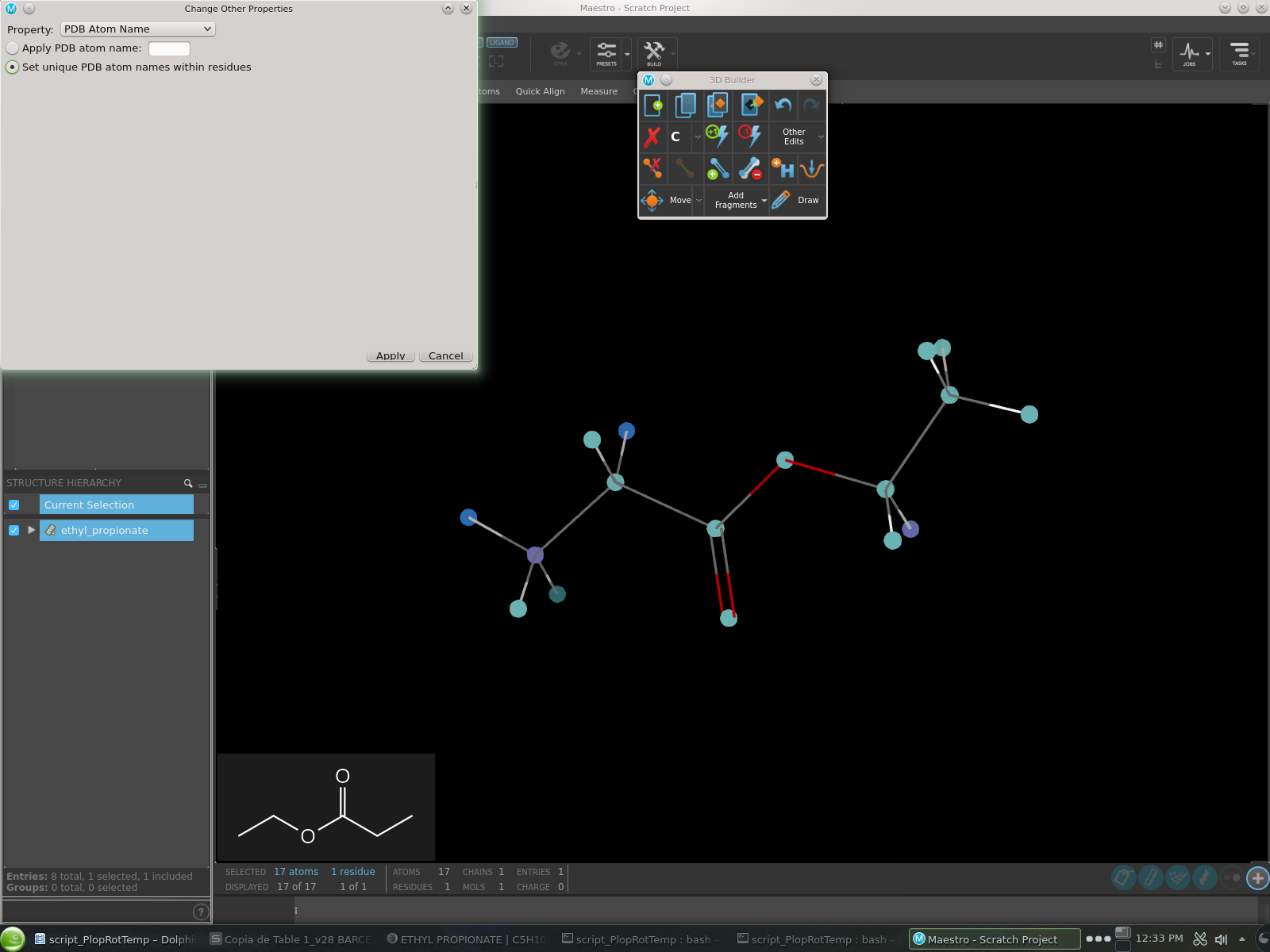
Fig. 12 Dialog to set unique PDB atom names for your ligand in Maestro.¶
Once you have your ligand in a MAE file, run:
$ /opt/schrodinger/utilities/python /opt/pele/scripts/PlopRotTemp.py -OPLS=2005 myligand.mae
If you have assigned special charges to your ligand (for example through a quantum calculation), then you can add the option -mae_charges=yes to preserve those charges.
Adding the generated template and rotamer library files to PELE¶
Say your ligand is named with the PDB code LIG. then, your files will be named by PlopRotTemp.py as:
ligLIG.rot.assign
If you want to add them to your PELE project’s ./DataLocal/ directory, remember to add a z at the end of the template file (which, in the example, will be named ligz). Place the template file under ./DataLocal/Templates/OPLS2005/HeteroAtoms/. If you are updating the central database, use Data/ instead of ./DataLocal/. As for the rotamer file, place it inside ./DataLocal/LigandRotamerLibs/; as before, use Data/ instead of ./DataLocal/ if you are updating the central database.
Usage documentation for PlopRotTemp.py¶
$ python /path/to/scripts/PlopRotTemp.py
$Revision: 1.13 $
For ligands:
Reads in a maestro mae file and makes a "rotamer enabled" template and the rotamer library to accompany it. This consists of finding the backbone core that results in the least number of child bonds rotated with any rotatable bond rotation. Reads in the rotatable bonds from a macromodel atomtyping (should be easily modifyiable to read them from stdin). Hetgrp_ffgen is used for atomtyping and to determine the geometry in the template fromt the mae. The mae must only have the residue to atomtype in it.
Builds rotamer libraries for arbitrary ligand molecules by building and combining rotamer libraries. There are two general algorithms that are implemented. This first is using macromodel (or some other external tool) to sample the degrees of freedom and converting the resulting ensemble into a rotamer library. The second is identifying the rotatable bonds, again using macromodel, and assigning rotamer libraries to these bonds. For most bonds a simple freely rotatable library ( 0,10,20,30...360), but in the case of closed rings special libraries are built using macromodel sampling. These component rotamer libraries are then arranged into groups for use in PLOP. Each group consists of a single tree rooted at the central core. This core can either be used chosen, or will be chosen based on an algorithm that minimizes the number of bond lengths from the farthest leeaf to the trunk. Any built rotamer libraries are placed in the current directory and a file called <resname>.rot.assign is also written. This tells PLOP how to assemble the full combinatoric library that will be used in sidehchain prediction/monte carlo.
For unnatural amino acids:
Requires: 1) a maestro mae file of an unnatural amino acid with no NMA or ACE termini; the N-H and C=0 are to be left as they are found in a peptide
2) a template file (specified by -t=<FILENAME>) created by hetgrp-ffgen using a maestro mae file of an unnatural amino acid with NMA or ACE termini present
Option Settings Required: -unnat=yes -t=<FILENAME> [other options] <RESIDUE_WITHOUT_CAPPING_GROUPS>.mae
Outputs: 1) a re-ordered template file (old one is backed up in FILENMAE.hetgrp_ffgen)
2) a PLOP nonstandard residue specification for pasting into a PLOP control file, both to stdout and to <maefile>_torsions.txt
Options
-core=<an1> Give one atom of the core section
-f=<an1>,<an2> Fix this bond number in the mae file.
CANNOT USE MACORMODEL SAMPLING WITH THIS OPTION
-tor=<an1>,<an2> Define torsions instead of using macromodel to do it.
CANNOT USE MACORMODEL SAMPLING WITH THIS OPTION
YOU MUST ALSO SPECIFY WHETHER THERE ARE RINGS OR NOT (-r)
-btor=<an1>,<an2> Define torsions for the backbone
-m=<yes/no> Wheter to split into multiple libraries
-min=<yes/no> Whether to minimize
-r=<yes/no/only> Whether to allow flexible rings
-t=<FILENAME> Use this original template instead of creating one
-o=<FILENAME> Write output to this filename (default is the residue name)
-g=<GRIDRES> Input the grid resolution for plop rotamer library
-goh=<GRIDRES> Input grid resolution for plop rotamer library (OHs)
-OPLS=<2001,2005> OPLS version to use
-mae_charges=<yes/no> Use mae charges for template file
-mtor=<number> Gives the maximum number of torsions allowed in each
group. Will freeze bonds to extend the core if
necessary.
-clean=<yes/no> Whether to clean up all the intermediate files
-d Debuging mode, assume all external programs have been
already run and the intermediate files were not cleaned
Options for Macromodeling Sampling
seting any of these options will do the sampling in macromodel to build
a sidechain library. Otherwise a series of libraries will be indentified
to be combined together in plop later.
-c=<FILENAME> Use the following pdb/mae file as an exhaustive list of
all confomers. This must be one file with multiple
models/entries. Still uses macromodel to find rotatable
bonds.
-n=<number> Maximum Number of Entries in Rotamer File
-a=<CGEN,MCMM...> Type of Search to Run for Sidechains, if none is specified
it will use PLOP sampling
-ba=<CGEN,MCMM...>Type of Search to Run for Backbones, if none is specified it
will not be sampled and a backbone library and no backbone
library will be created
-s=<number> Maximum Conformers to Sample
-e=<number> Energy Cutoff (in kJ/mole)
-md=<number> Maximum distance between atoms in equivalent struct(A)
Options for Unnatural Amino Acids
-chain=<chain> For output of PLOP-style nonstandard residue specification,
set the chain name equal to what it would be in the whole
macromolecule
-res=<num> For output of PLOP-style nonstandard residue specification,
set the residue number equal to what it would be in the whole
macromolecule
Mae file should be properly atomtyped
Most common problem: As part of this procedure the pdb atom names are often renamed when they are not unique. Alsothis procedure works best if the ligand is minimized. For these two reasons an atomtyped, minimzed version of the input ligand is written to (input).PlopRotTemp.pdb. If at all possible, use the ligand structure and atom names from this file in any subsequent plop runs.
examples:
Build a rotamer library for the following ligand at a grid resolution of 20 deg using PLOP/PRIME to combine libraries
$SCHRODINGER/utilities/python PlopRotTemp.py 3ert_lig.mae -g=20
Build a rotamer library for the following ligand at using CGEN sampling in macromodel.
$SCHRODINGER/utilities/python PlopRotTemp.py 3ert_lig.mae -a=CGEN
Build a rotamer library for the following ligand at a grid resolution of 20 deg using macromodel to sample any rings and combining this with freely rotatable libraries in PLOP to create combined libraries for the ligand.
$SCHRODINGER/utilities/python PlopRotTemp.py 1rth_lig.mae -r=yes
Make libraries for rotatable bonds in ligand.mae up to a maximum of 4 rotatable bonds in each library
All additional bonds are defined as backbone and are sampled with CGEN to produce a backbone libary
$SCHRODINGER/utilities/python PlopRotTemp.py ligand.mae -mtor=4 -ba=CGEN
For a given ligand named LIG the following files will be created:
lig - Template file for use in PLOP, its zmatrix matches the libraries created
LIG.rot.assign - Summary of all libraries build or used for this ligand read into plop with the command
"rot assign all"
LIG???.side - (OPTIONAL) Component sidechains libraries created if there are closed rings or CGEN sampling is used
LIG__B.back - (OPTIONAL) Backbone sidechain library
---------------------------------------------------------------------------------------------------------
All jobs run on the localhost
usage: "$SCHRODINGER/utilities/python PlopRotTemp.py [file.mae]"
AMBER¶
A quick way to create a template file for any of the AMBER force fields used in PELE is by using the scripts/transformLigandOPLSTemplateToAmber.py script that comes with PELE. Notice that this script needs an existing OPLS template file to work, and that it uses exactly the same parameters as in OPLS, but transforms them as needed by AMBER. This only affects the radii, which are expressed as diameters in OPLS, but as radii in AMBER. The OPLS parameters may, or may not, work for your system, so proceed with caution.
To create the AMBER template for your ligand template lig (without the ending character z), run the script as:
$ python /path/to/pele/scripts/transformLigandOPLSTemplateToAmber.py lig
You will find your AMBER template as amberTemplatesResults/ligz.
Hetero group rotamer library files¶
See also Ligand rotamer library file for information on the format of the generated file.
If you have the template file for your molecule, but not the rotamer file, you can create it using, as before, PlopRotTemp.py (see OPLS). Notice that, since PlopRotTemp.py works with OPLS, your template file may need to be an OPLS template file (though the rotamer file will be useful for any force field). You will still need as input a MAE file. The output will be, again, a template file with the name of the ligand (this will be a copy of the input template file), and a rotamer file.
$ python /path/to/scripts/PlopRotTemp.py -t=myligand.template myligand.mae
As before (for the template and rotamer files generation), you can place your rotamer file inside ./DataLocal/LigandRotamerLibs/; use Data/ instead of ./DataLocal/ if you are updating the central database.
Conformation library files¶
See also Conformation library file for information on the format of the generated file.
To create a conformation library file you need to use the peleffy package and have the output of a run of the Bioactive Conformational Ensemble (BCE) server. If we assume that peleffy is installed and the output of the BCE server is in BCE_output and the ligand structure is in input.pdb, the dihedral library could be generated using the following command:
$ python -m peleffy.main input.pdb --conformations_info_path BCE_output/ --as_datalocal -c opls2005 -f OPLS2005
This command would generate a DataLocal folder with the conformation library inside the Conformations subfolder, as well as the OPLS template for the ligand. For more information about peleffy visit their documentation.