MD¶
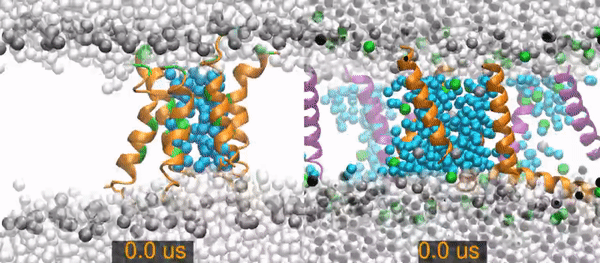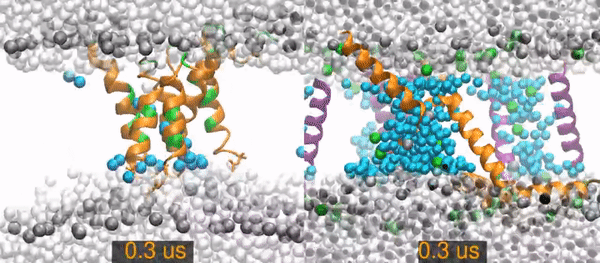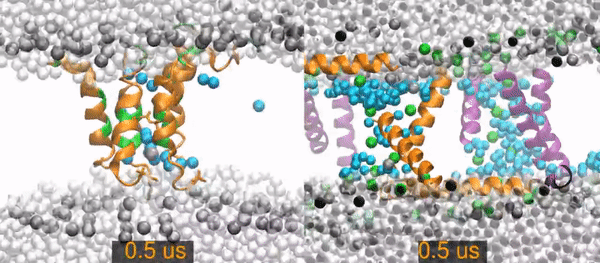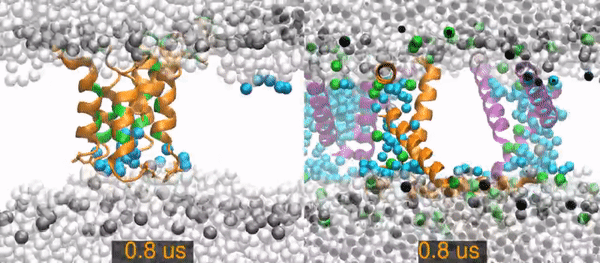
MD Tutorial:¶
- Raw Input preparation
Open maestro preprocess the structure (capping, protonation, tautomers…) and make sure you do not have any mistake on the output structure.
- Processed input preparation
Save 1 pdb called complex.pdb with receptor-ligand and structural water molecules
Save 1 mol2 called ligand.mol2 with the already protonated ligand
- Ligand Preparation
export AMBERHOME=/shared/opt/AmberTools/amber16/
$AMBERHOME/bin/antechamber -i input_name.mol2 -fi mol2 -o output_name.prep -fo prepi -nc charge_of_ligand -c charge_model -at atom_types -j processors_to_use
i.e $AMBERHOME/bin/antechamber -i ligand.mol2 -fi mol2 -o ligand.prep -fo prepi -nc -1 -c bcc -at gaff -j 4
- Receptor Preparation
$AMBERHOME/bin/pdb4amber -i input_name.pdb -o output_name.pdb
i.e $AMBERHOME/bin/antechamber -i complex.pdb -o complex_processed.pdb
Read the output and make sure there are no errors in your input
- Parametrize ligand
$AMBERHOME/bin/parmcheck2 -i ligand.prep -f prepi -o ligand.frcmod
- AmberTools input
cp /shared/data/software/MD/leap.in your_directory
fill in the fields indicated on the file
- Build Topology & Coordinates file
leap -sf leap.in
check the warning outputs and remove the hydrogens from the complex_processed.pdb that they contain clash (as they will be automatically added by tleap) fix other errors.
lauch tleap again
- Visualize the ouput complex.pdb and make sure everything looks alright
protein is inside the box
ligand looks normal