Structure Filtering Tutorial¶
This tutorial aims to guide the user to perform a database filtering with the possibility of specifying which R-groups we want fixed.
Flags and Input Files¶
-i TEMPLATE_LIGAND, –template_ligand –> Path to PDB template ligand.
-l LIGANDS, –ligands –> Path to SDF file with database ligands or folder with SDF files.
-o OUTFILE, –outfile –> Output file name.
-a ATOM_LINKER, –atom_linker –> PDB atom name of core that is bound to R-group.
Requirements of the input files¶
The file of the substructure we want to have in all the molecules, must be in PDB format and have unique PDB atom names.
The database files must be SDF format.
Example - Running in a Jupyter Notebook¶
import database_filtering
from database_filtering.utils.utils import filter_mols
import rdkit
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import rdMolDraw2D
from rdkit.Chem.Draw import IPythonConsole
IPythonConsole.molSize = 300,300
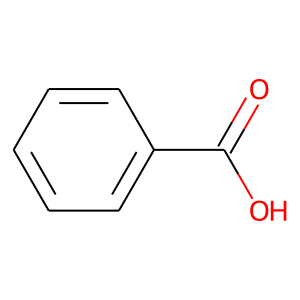
template_ligand = Chem.MolFromSmiles("O=C(O)c1ccccc1")
template_ligand
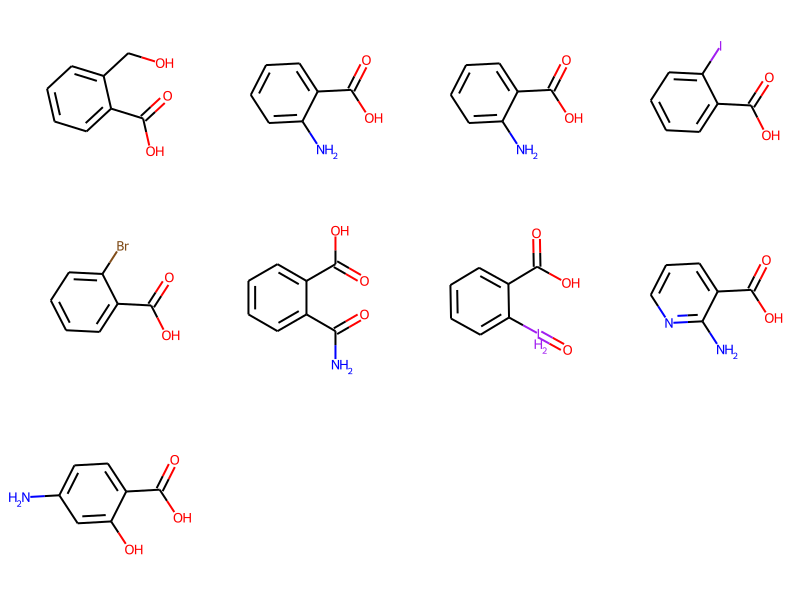
ligands = [Chem.MolFromSmiles("O=C(O)c1ccccc1CO"),
Chem.MolFromSmiles("Nc1ccccc1C(=O)O"),
Chem.MolFromSmiles("Nc1ccccc1C(=O)O"),
Chem.MolFromSmiles("O=C(O)c1ccccc1I"),
Chem.MolFromSmiles("O=C(O)c1ccccc1Br"),
Chem.MolFromSmiles("NC(=O)c1ccccc1C(=O)O"),
Chem.MolFromSmiles("O=[IH2]c1ccccc1C(=O)O"),
Chem.MolFromSmiles("Nc1ncccc1C(=O)O"),
Chem.MolFromSmiles("Nc1ccc(C(=O)O)c(O)c1")]
Draw.MolsToGridImage(ligands,molsPerRow=4,subImgSize=(200,200))
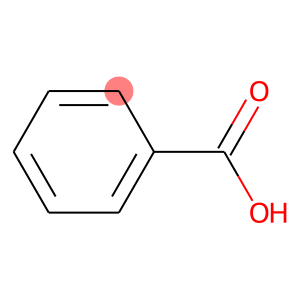
linker = ['C7']
template_ligand.__sssAtoms = [8] # Highlight the atom C7
template_ligand
REMEMBER: If your linker is atom C7, we will only obtain molecules that have an R-group bound to that atom.
Also, we can select more than one linker atom:
In a notebook --> linker = ["C7", "C4"]
Running on the cluster --> -a C7 C8
template_ligand_path = "./template_ligand.pdb"
ligands_path = "./ligands.sdf"
filter_mols(template_ligand_path, ligands_path,'test',linker)
# Results are stored in the file test.sdf
Filtering passed for molecule
Filtering passed for molecule
Filtering passed for molecule
Filtering passed for molecule
Filtering passed for molecule
Filtering passed for molecule
Filtering passed for molecule
No substructure match for ligand , skipping
Molecule did not meet the R-groups requirements.
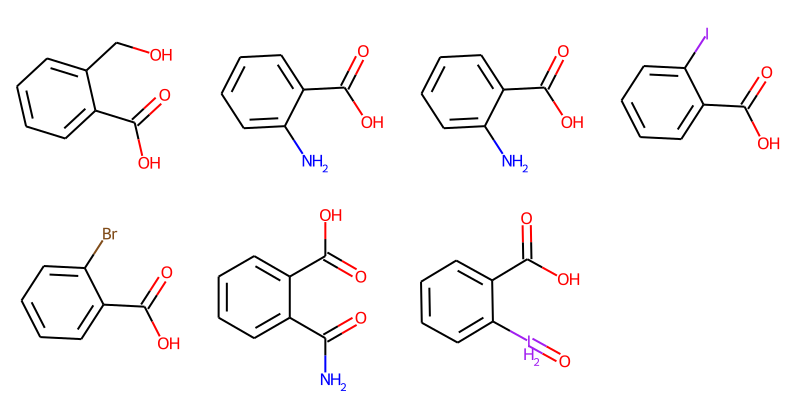
Filtering Results¶
Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200))
Running in the Cluster¶
#!/bin/bash
#SBATCH -J filter
#SBATCH --output=filter.out
#SBATCH --error=filter.err
#SBATCH --ntasks=3
#SBATCH --mem-per-cpu=10000
source /shared/home/hmartin/miniconda3/etc/profile.d/conda.sh
conda activate /shared/home/hmartin/miniconda3/envs/r_groups_env
python -m database_filtering.run_filtering -i template_ligand.pdb -l ligands.sdf -a C7 -o test