Drug discovery¶
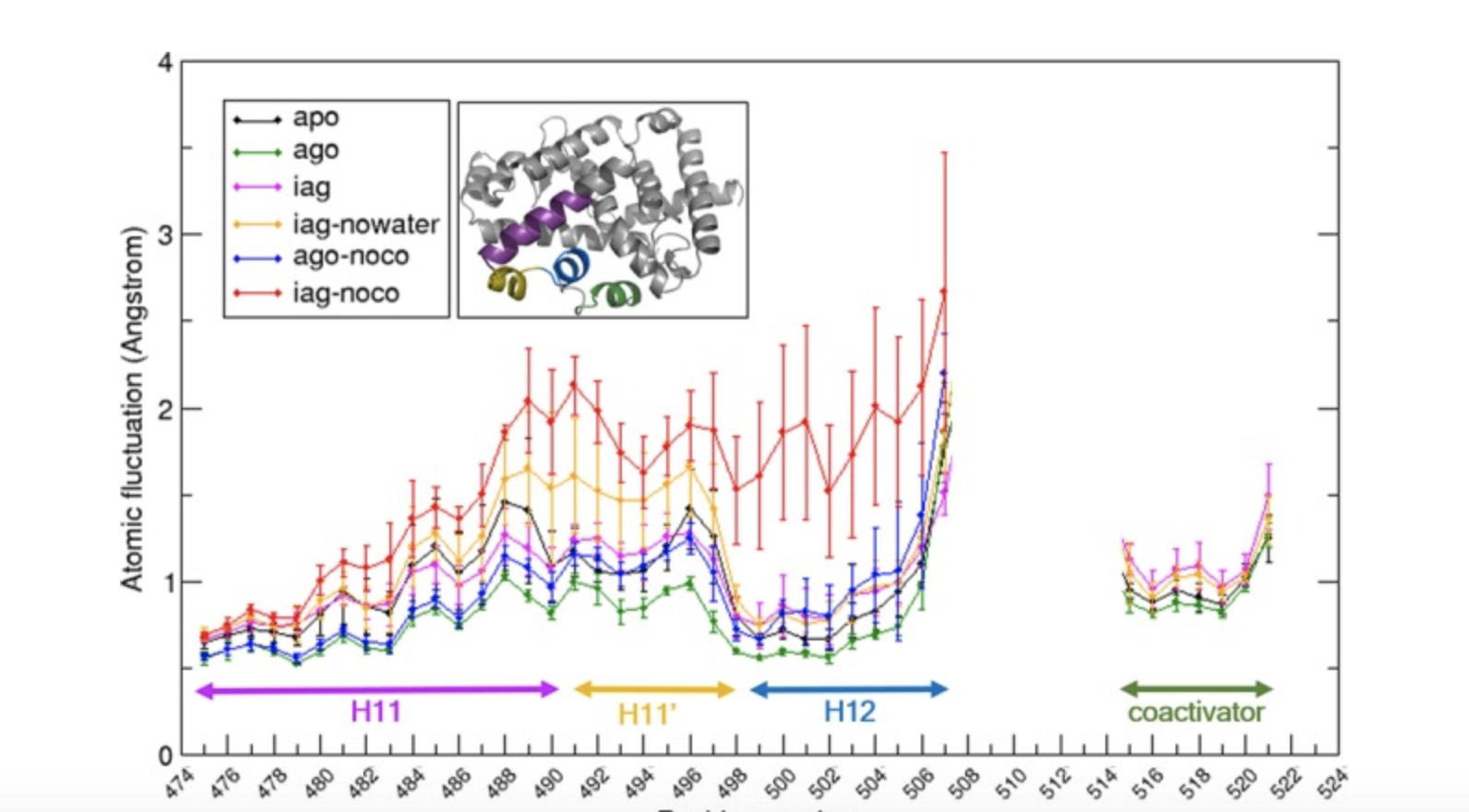
Atomistic Simulations Shed New Light On The Activation Mechanisms Of RORγ And Classify It As Type III Nuclear Hormone Receptor Regarding Ligand-Binding Paths¶
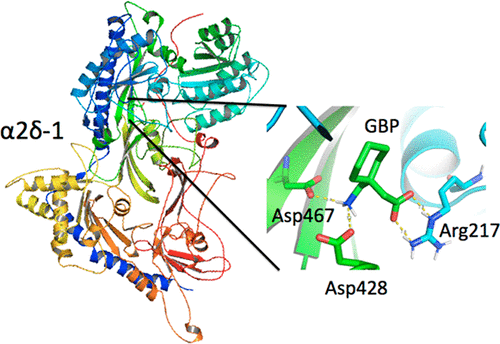
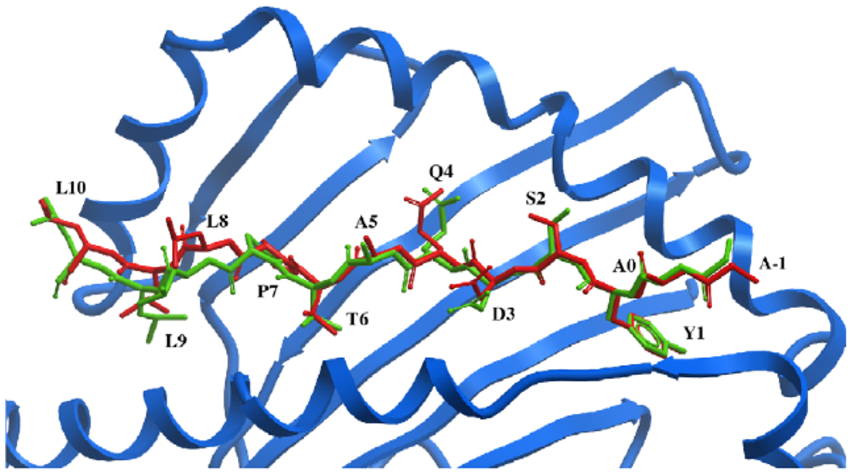
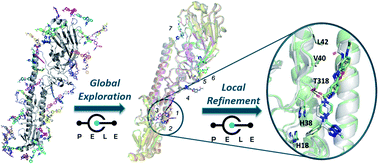
Pushing The Limits Of Computational Structure-Based Drug Design With A Cryo-EM Structure: The Ca2+ Channel α2δ-1 Subunit As A Test Case¶
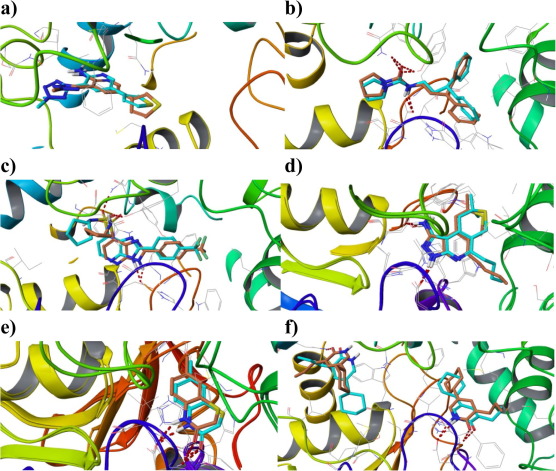
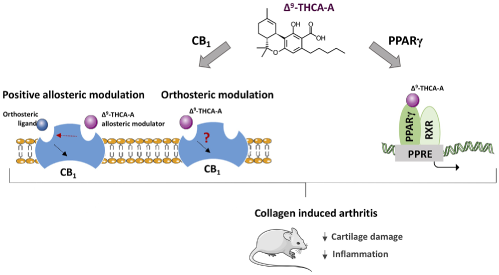
Δ(9)-Tetrahydrocannabinolic Acid Alleviates Collagen-Induced Arthritis: Role Of PPARγ And CB(1) Receptors¶