Hemagglutinin: Pocket Detection¶
3- Flu hemagglutinin: It is one of the two proteins found on the surface of the influenza viruses. Hemagglutinin A (HA) is an antigenic glycoprotein assembled as a homo-trimer of identical subunits that are formed of two disulfide-linked polypeptides: membrane-distal HA1 and the smaller, membrane-proximal, HA2. HA is responsible for host cell binding and subsequent fusion of viral and host membranes in the endosome after the virus has been taken up by endocytosis.
Cyclic and linear inhibitors that bind to the surface of hemagglutinin have been identified. Structural information on the antibody-bound target is also available (Fig. 1).
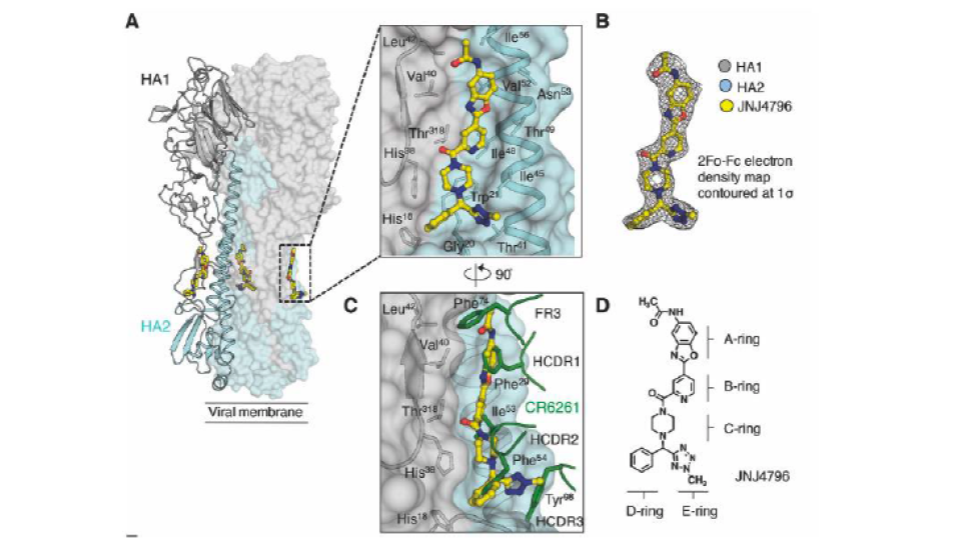
Fig. 4 Hemagglutinin system¶
Objective¶
The goal is to predict the binding mode of JNJ4796 to a structure of hemagglutinin which is not in a conformation likely to bind JNJ4796. For this, the structure found in PDB entry 5W6T (bound to a cyclic peptide) was taken for induced fit PELE simulations. The question to be answered is: Can PELE find the binding site and then the correct binding mode by doing a 2-step procedure (global exploration + local refinement)?
Preparation¶
Receptor¶
5W6T processed with Protein Preparation Wizard (Schrodinger). Of note, no crystal structure of the protein with JNJ4796 bound is available in the pdb, so the reference interactions were defined in terms of distance to the residues that are pictured in Fig 1.
Procedure: PELE Global Exploration¶
Main simulation characteristics¶
Starting poses for PELE Local Exploration: 40 initial poses at random positions around the whole protein were generated (Fig. 2)
“inverselyProportional”: Emphasize clusters with low representation
Iterations: 100
PeleSteps: 4
Processors: 250
Compute time: 12-15h
ControlFile specifics¶
system: 'input_0.pdb' #Input ligand-protein pdb
chain: 'L' #Ligand chain name
resname: 'JNJ' #Ligand residue name
seed: 12345
ppi: true #Run a global exploration + clustering + inducedfit
cpus: 100 #Number of cpus
atom_dist: #Check the distance between this atoms
- L:1:O3
- A:315:CA
Results¶
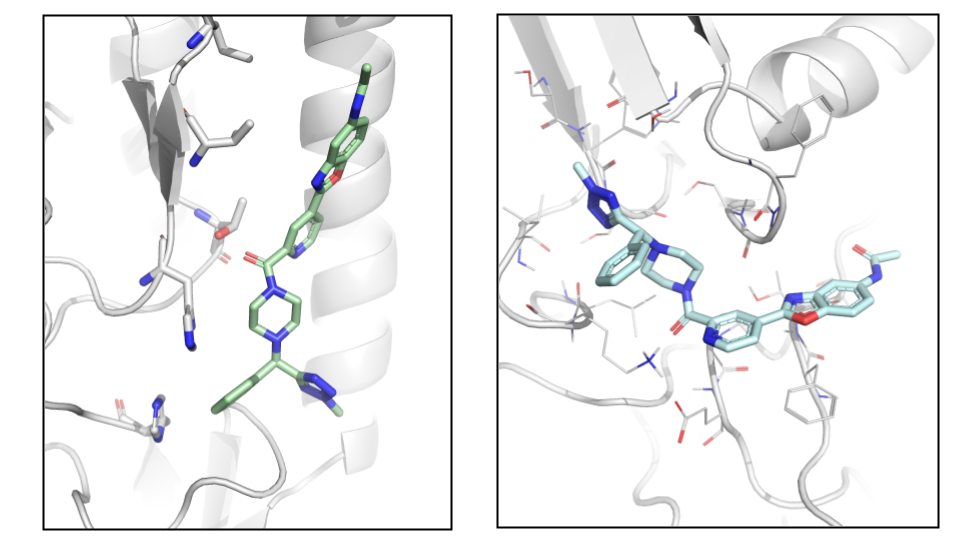
The energy profile for the refinement of the 7 poses can be found in Fig. 5. The two lowest energy minima are highlighted. The lowest energy pose with an H-bond to T318 (Fig. 6) is the one with the lowest interaction energy (-50 kcal/mol) as predicted by PELE (Fig. 5). This pose is very close to the binding mode described by Kadam et al., 2017.
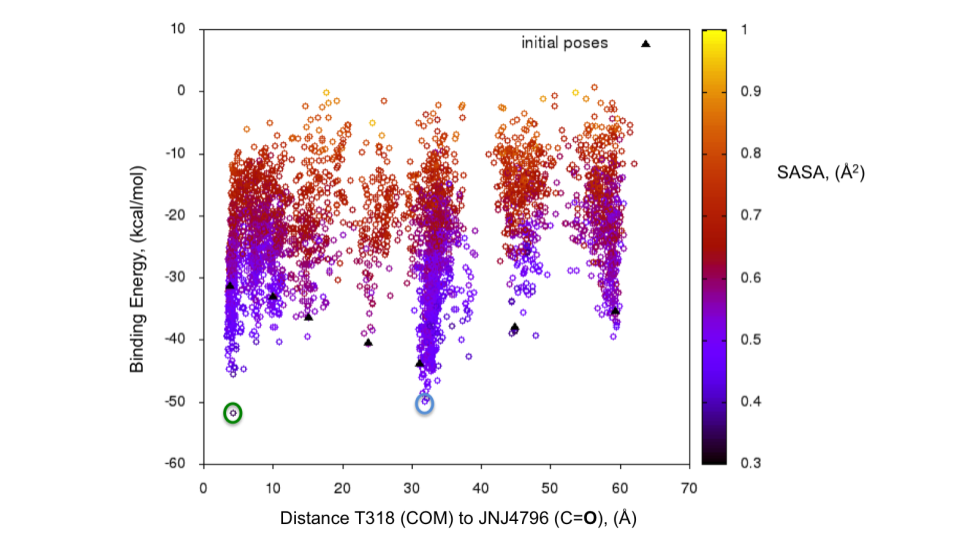
Fig. 5 Binding energy plot against distance to T318 (one of the key residues anchoring JNJ4796) for the 7 poses generated with the initial global exploration. The two minima which are highlighted with green and cyan circles are the ones represented in Fig. 7 below.¶