Frequently asked questions (FAQs)¶
If you do not find a solution to your problem, feel free to send us an email at pelesupport@nostrumbiodiscovery.com. We’re here to help!
1. Why does it ask me to check atoms?¶
Example error message¶
The following error message indicates that the atom(s) you chose to calculate the distance metric or constrain does not exist.
Check the atoms A:999:C given to calculate the distance metric.
Solution¶
To resolve the issue, you should:
ensure the atom string has the correct format, i.e.
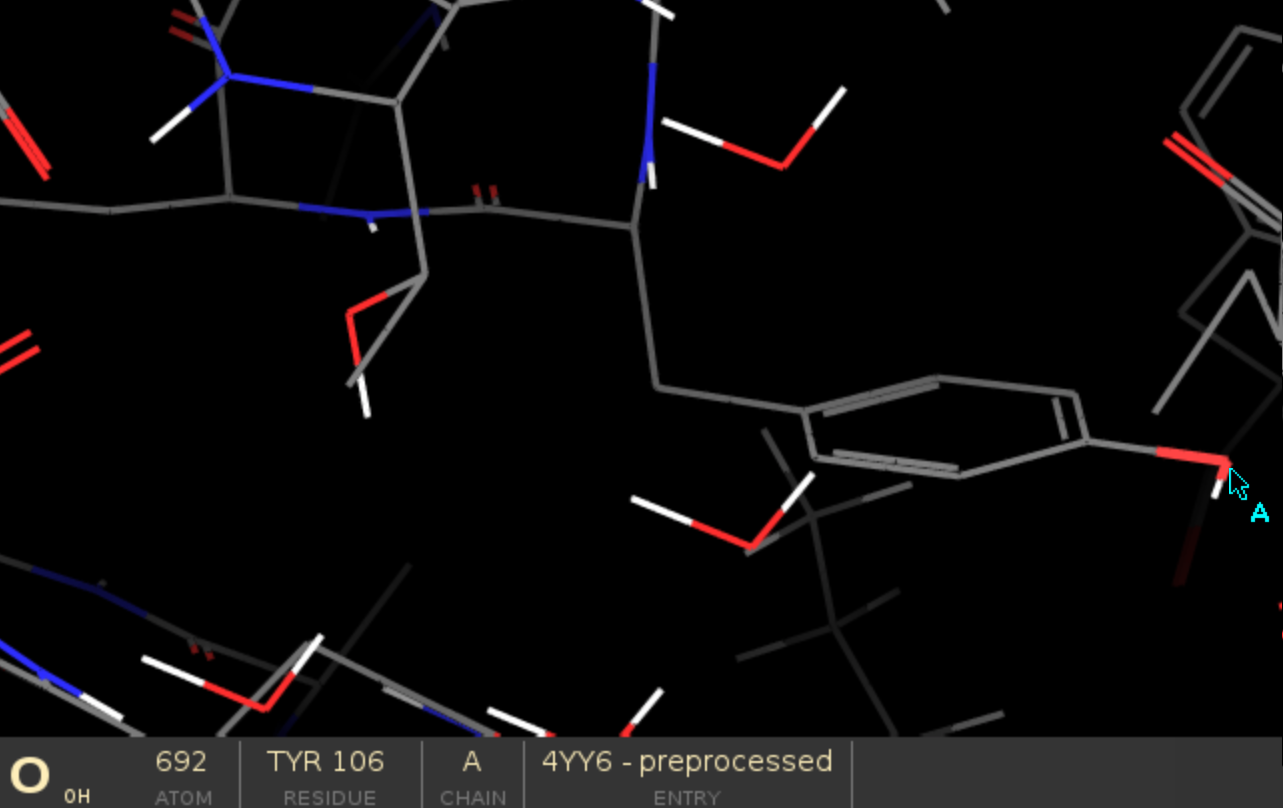
"chain_id:residue_number:atom_name"examine the protein structure in Schrödinger Maestro (or open the PDB file as text) and double-check, if the atom exists.
You can easily check residue numbers and atom names in the bottom panel in Maestro by hovering the mouse pointer over a specific atom. In this example, the correct atom string would be "A:106:OH".
2. PELE cannot locate a file, how do I fix it? (FileNotFoundError)¶
Example error message¶
FileNotFoundError: [Errno 2] No such file or directory: '/home/anna/errors/file.pdb'
Solution¶
If PELE raises FileNotFoundError, it probably means it cannot find one of the files specified in input.yaml such as system, ligand or RMSD reference. Make sure:
there are no spelling mistakes in file names
all required files are located in your working directory (or provide a relative path in your input file instead).
3. Why did it fail to extract the ligand? (ValueError)¶
Example error message¶
ValueError: Something went wrong when extracting the ligand. Check residue&Chain on input
Solution¶
This error indicates that the software was not able to find the ligand in the PDB file. Make sure chain and resname flags
in your input file have correct values. Remember that the ligand needs to have a unique chain ID!
4. How to add CONECT lines? (ConnectionsError)¶
Example error message¶
pele_platform.Errors.custom_errors.ConnectionsError: Your PDB file is missing the CONECT lines. Please do not remove them after Schrodinger preprocessing.
Solution¶
This warning indicates that the PDB file is missing the connectivity section. To resolve the issue, you should import the PDB in Schrödinger Maestro and preprocess it, launching the Protein Preparation Wizard.
Note that CONECT lines are required for peleffy forcefield builder.
5. How to handle ligand parametrization failure?¶
Example error message¶
Sometimes parametrization of a hetero molecule (cofactor, modified residue, crystallization factor) will fail, which should result in the following warning message:
Failed to parametrize residue MET. You can skip it or parametrize manually
(see documentation: https://nostrumbiodiscovery.github.io/pele_platform/errors/index.html#parametrization).
The error raised was: Size of atom parameter lists should match.
Solution¶
Depending on the complexity of the system you are studying, there are two options available:
a. Remove the residue¶
If the hetero molecule is not necessary to study your system, the easiest way to handle this is to remove it from the PDB file. Similarly, you can ignore the warning, but PELE is likely going to crash because it will miss the template.
b. Parametrize manually¶
Alternatively, you can parametrize the molecule manually and pass obtained template and rotamer files in the input.yaml.
Save the residue to a separate PDB file, ensure the CONECT lines are included and the Lewis structure is correct.
Run the following command inside the Python environment of the platform to create the default rotamer and template files. For more options, please refer to the Open Force Field for PELE documentation.
python -m peleffy.main ligand.pdb
Add paths to your newly created files to the input.yaml, for example:
templates: - "/path/to/metz" rotamers: - "/path/to/MET.rot.assign"
6. How to analyse the output files?¶
You should mostly focus on the results directory, which contains curated, user-friendly analysis stored in the following folders:
top_poses- top 100 lowest binding energy structures
plots- plots of multiple metrics selected by the user
clusters- lowest binding energy cluster representatives and clustering plots.
If you want to understand more about other files generated by the Platform, please refer to Understanding the output files tutorial.