PDB preparation¶
This section contains instructions and recommendations to help you with the preparation of the input PDB files that PELE needs in order to run successfully.
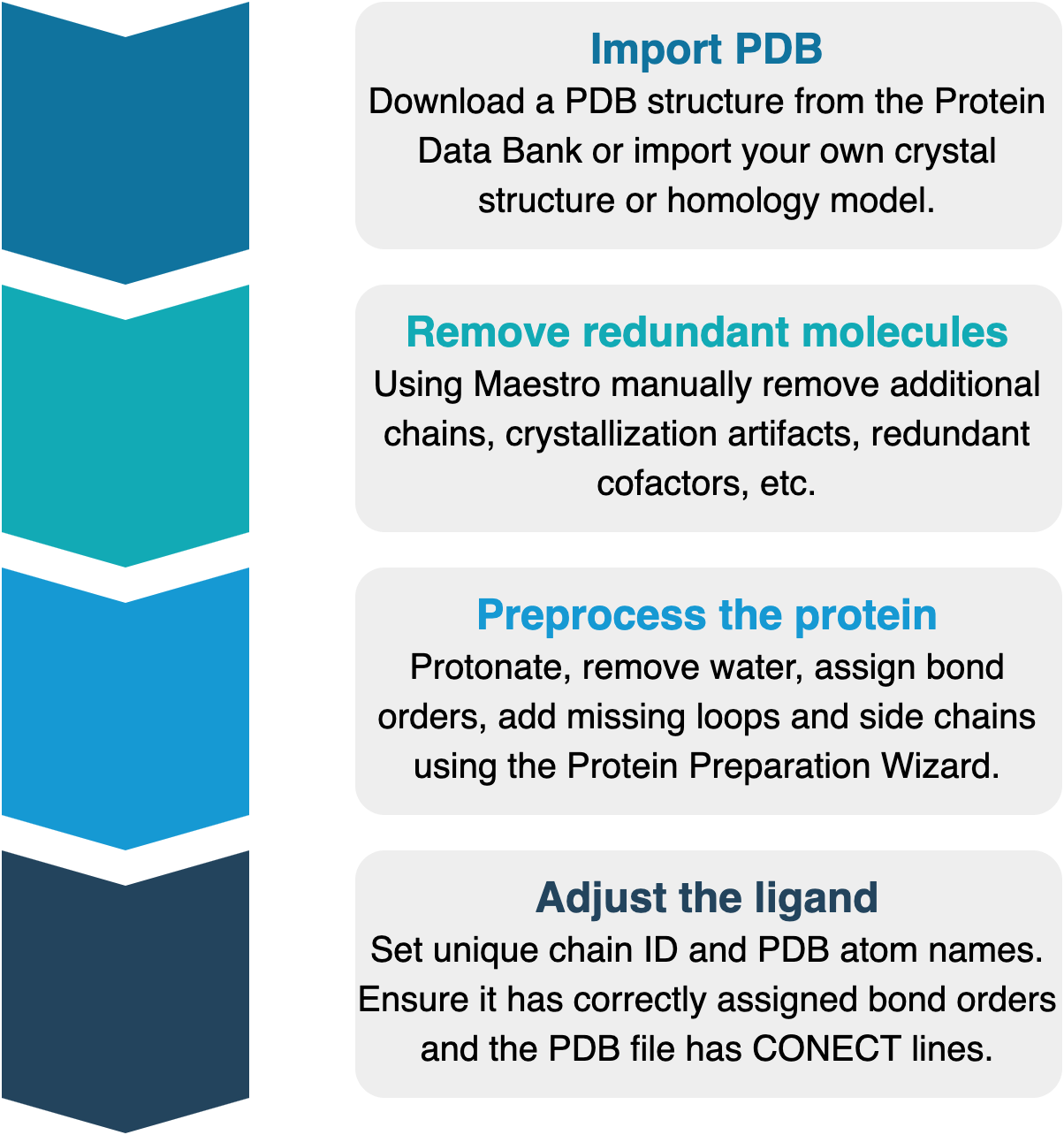
Protein preprocessing workflow using Schrödinger tools¶
We have a collection of tutorials where we explain step by step the procedure to prepare the initial structure for PELE.
Import PDB¶
We normally start from a crystallographic structure of the system that we want to simulate. Crystals can belong to either holo or apo forms of our target. Structures obtained with homology modelling can also be used.
In case small backbone movements are required for a particular study (e.g. to host a ligand into a binding site where a close loop needs to be shifted), PELE’s algorithms will probably be enough to explore these rearrangements. When big conformational movements on the protein backbone are pursued, molecular dynamics simulations might be a good strategy to produce different protein conformations from where PELE simulations can start. In any case, we must ensure that our model is as accurate as possible since the success of our simulation will depend on it.
Remove redundant molecules¶
Heteromolecules and waters¶
It is worth checking the necessity and importance of cofactors, metal ions, water molecules, crystallization agents, etc. Particularly, we might consider removing most of the water molecules that are present in our structure. PELE uses an implicit solvent to account for its effects. Thus, we normally do not need to keep them. However, there are mainly three exceptions:
Structural water: water molecules inside protein cavities that are essential for the stability of the protein must be kept. These water molecules are usually participating in 3 or 4 hydrogen bonds with the protein.
Coordination water: water molecules coordinated with metals must be kept into the structure to satisfy the coordination geometry around the metal center. In this case, the Platform will automatically constrain these water molecules around the metal.
Interfacial water: water molecules that mediate with protein-ligand interactions can also be included explicitly. In this case, we should use the aquaPELE algorithm. Please, refer to aquaPELE parameters to learn how to activate it.
Redundant chains¶
It also makes sense to remove additional protein chains to speed up the simulation. However, if a protein belongs to a biologically active dimer, we normally will have to include both monomers to the simulation. Especially, if the region of interest lies in the interface between both monomers.
Preprocess the protein¶
Fix missing loops¶
This is a matter of improving the quality of our model rather than a requirement of the Platform. However, it is extremely important to spend some time analyzing the validity of the structure that we want to simulate. It is recommended to add missing loops that are expected to be near the region of interest of our system. There are several strategies that can help us with this task like applying homology modelling on our structure or running computational tools to add those missing loops like Prime from Schrödinger.
Fix missing atoms/residues¶
The Platform will require the PDB to have all the atoms that standard protein residues are expected to have (according to the internal templates of PELE). For example, hydrogen atoms typically need to be added to our structure. This task can be easily done with Maestro from Schrödinger. There is a tool called Protein Preparation Wizard that is able to add missing atoms to standard residues and assign their proper atom names. In most cases, the Platform will be able to run with systems that have been prepared with this tool.
Check protonation state¶
To obtain a reliable model, we must ensure to have the right protonation states. Tools like PROPKA or the H bond optimizer in Protein Preparation Wizard can be very helpful.
Heteromolecule parametrization¶
Any non-standard residue present in the input structure must be parametrized before running a PELE simulation. The Platform takes care of this task automatically. However, to avoid any problems during the parametrization process, it is recommended to assign the right connectivity to any heteromolecule. We also must ensure that connectivity information is saved in the resulting PDB file through CONECT lines.
Please, check this tutorial if you want to learn more about heteromolecule parametrization.
Adjust the ligand¶
The ligand structure needs to meet a number of criteria in order to be correctly perturbed by PELE. Most importantly, it needs to have a unique chain ID and any residue name except for “UNK”. Additionally, it needs to have correctly assigned bond orders, including CONECT lines in the PDB file, as well as unique PDB atom names.
If you are not sure how to correctly prepare the ligand, you can follow section 1d from the Fragment growing tutorial.