Ligand parametrization using peleffy¶
Introduction¶
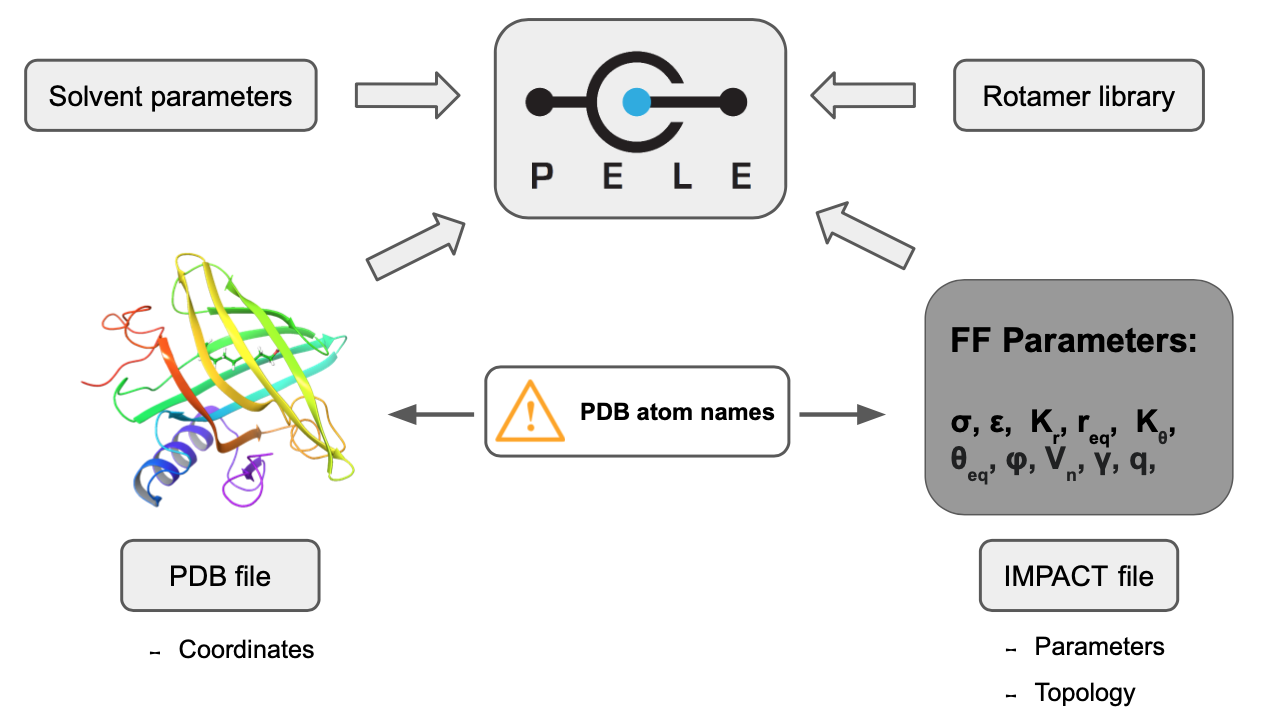
The main purpose of peleffy is to build the parameter files for PELE. Basically, PELE requires two files for each non-standard residue found in the system:
IMPACT template: a file containing the atom types and parameters of the ligand. Its job is to link each atom with the corresponding parameters using PDB atom names. Thus, PDB atom names in the input PDB file must match with the expected PDB atom names in the Impact file. This file intrinsically contains the information about the topology and connectivity of each residue.
Rotamer library: a file containing the branches that can rotate with respect to a central atomic core. Each branch consists in a set of consecutive rotatable bonds.
Besides, a third file with the Solvent parameters might be required when employing the OBC implicit solvent.
Prerequisites¶
Peleffy comes with the PELE Platform installation, so make sure you activate the correct python environment before attempting the tutorial, e.g.
conda activate pele_platform
You also might need Schrödinger software (any version or license type), if your ligand is not yet preprocessed.
Input files¶
You will need a PDB file containing the ligand, make sure it has:
unique PDB atom names
correct protonation
CONECT lines at the end of the file.
To ensure ligand atoms have unique PDB atom names:
Open your ligand file in Schrödinger
Select the ligand with a mouse click
Go to
Buildand click onOther edits -> Change atom propertiesSelect
PDB atom namefrom the drop down list and selectSet unique PDB atom names within residues.
If your PDB file is missing the CONECT lines:
Make sure you did not disable them in the export window of Maestro.
Check if your bond orders are assigned correctly, they can be adjusted with Protein Preparation Wizard tool.
Command line¶
The easiest way to parametrize your ligand is through the command line interface:
python -m peleffy.main my_ligand.pdb
If you need to tweak any parameters, you can choose from several command line arguments:
Parameter |
Argument |
Functionality |
|
None |
Show help message and exit |
|
string |
OpenForceField’s forcefield name. Default is openff_unconstrained-1.2.0.offxml. |
|
string |
Output path. Default is the current working directory |
|
integer |
Rotamer library resolution in degrees. Default is 30. |
|
None |
Generate solvent parameters for OBC. |
|
None |
Output will be saved following PELE’s DataLocal hierarchy. |
|
string |
The name of the method to use to compute charges, you can choose one from: gasteiger, am1bcc, OPLS. |
|
string |
The path to the file with charges. |
|
string |
Chain ID of the molecule to parameterize. |
|
None |
Do not exclude terminal rotamers when building the rotamer library. |
Example: Parametrization of ligand contained in my_ligand.pdb file with openff-1.3.0 force field, rotamer resolution of 10 degrees, am1bcc charge calculation method and custom output directory.
python -m peleffy.main my_ligand.pdb -f openff_unconstrained-1.3.0.offxml -o "ligand_params" -r 10 -c am1bcc
API¶
If you are comfortable with python, you can make use of peleffy’s API to parametrize your ligand. Follow the steps below to get a general idea how it works and for more details, please refer to peleffy documentation.
The first step is always initializing a Molecule object, you can do it either from a PDB file or a string of SMILES
from peleffy.topology import Molecule
molecule = Molecule("my_ligand.pdb") # from a PDB file
molecule = Molecule(smiles='c1ccc2cc3ccccc3cc2c1', hydrogens_are_explicit=False) # from SMILES
Generating rotamer library
# Create a rotamer library
rotamer_library = RotamerLibrary(molecule)
rotamer_library.to_file('LIG.rot.assign')
Parametrization and creating the Impact file
with OpenFF
from peleffy.forcefield import OpenForceField
from peleffy.topology import Topology
from peleffy.template import Impact
openff = OpenForceField('openff_unconstrained-1.2.0.offxml')
parameters = openff.parameterize(molecule)
topology = Topology(molecule, parameters)
impact = Impact(topology)
impact.to_file('ligz')
with OPLS2005
from peleffy.forcefield import OpenForceField
from peleffy.topology import Topology
from peleffy.template import Impact
openff = OpenForceField('openff_unconstrained-1.2.0.offxml')
parameters = openff.parameterize(molecule, charge_method='OPLS2005')
topology = Topology(molecule, parameters)
impact = Impact(topology)
impact.to_file('ligz')
Generating solvent parameters
with OpenFF
from peleffy.solvent import OBC2
solvent = OBC2(topology) # use previously generated topology
solvent.to_file('ligandParams.txt')
with OPLS2005
from peleffy.solvent import OPLSOBC
solvent = OPLSOBC(topology) # use previously generated topology
solvent.to_file('solventParamsHCTOBC.txt')
Templates usage¶
Once the templates have been generated, they can be used in a PELE simulation. PELE will need these templates for any non standard residue included in the system to simulate. It is important to mention that PDB atom names of each non standard residue in the system must match with those names from the structures employed in the template generation. A good practice is to assign unique PDB atom names to non standard residues present in the system, isolate them and, one by one, generate their templates with peleffy. In this way, the system that will be simulated will already contain non standard residues with the right PDB atom names.
Templates must be saved in specific locations so PELE can find them. Each type of template must be saved in a different folder inside the DataLocal directory. DataLocal must be created in the working directory where the simulation runs.
Template |
Filename |
Directory |
OPLS2005 parameters template |
ligz, resz, xxxz |
|
OpenFF parameters template |
ligz, resz, xxxz |
|
Rotamer library |
LIG.rot.assign, RES.rot.assign, XXX.rot.assign |
|
OBC solvent template (OPLS2005) |
ligandParams.txt |
|
OBC solvent template (OpenFF) |
solventParamsHCTOBC.txt |
|